iVAE documentation
iVAE is an enhanced representation learning method designed for capturing lineage features and gene expression patterns in single-cell transcriptomics. Compared to a standard VAE, iVAE incorporates a pivotal interpretative module that increases the correlation between latent components. This enhanced correlation helps the model learn gene expression patterns in single-cell data where correlations are present.
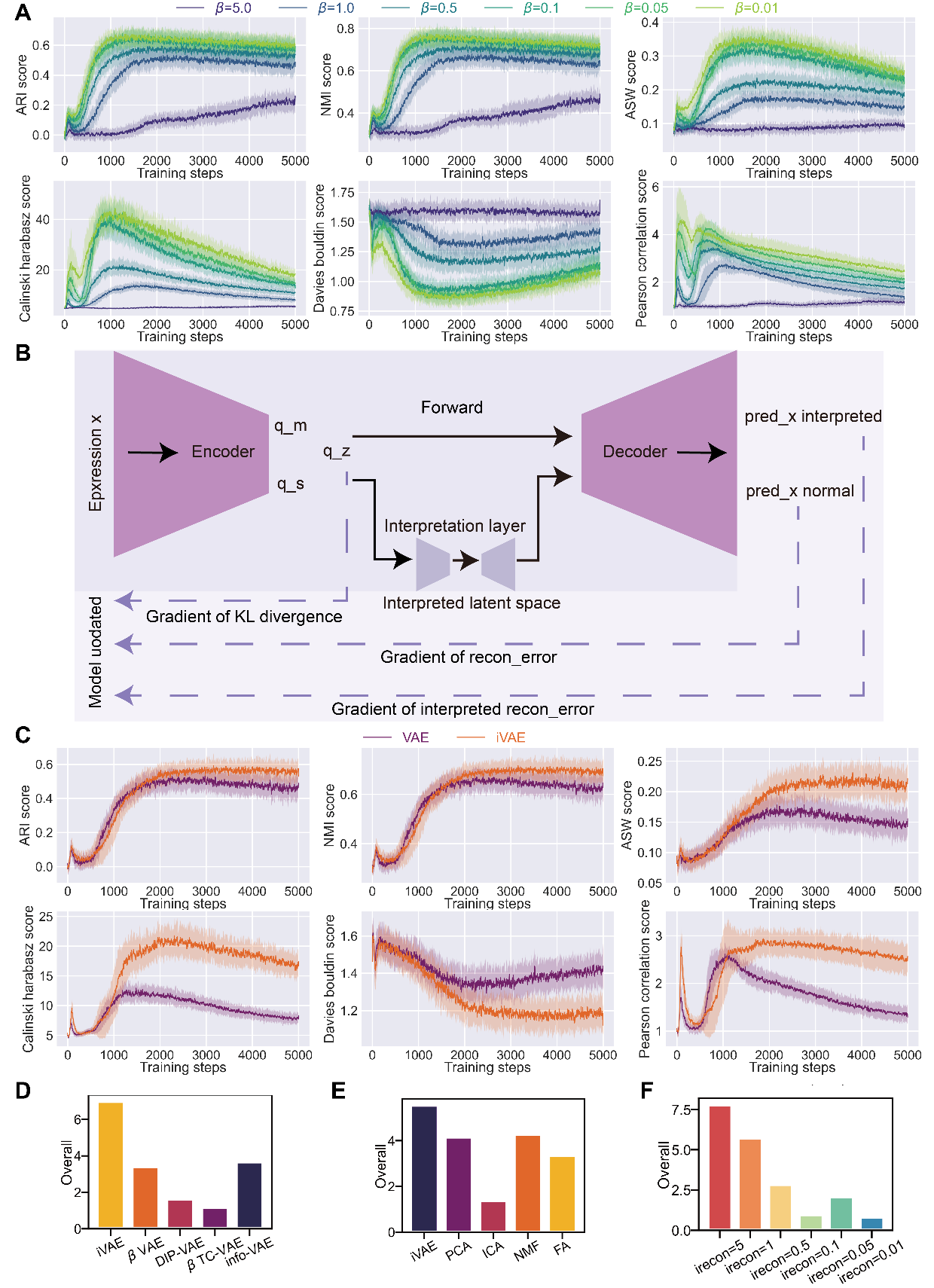
Key Features
Interpretative Module: A bottleneck layer that compresses and reconstructs the latent space, revealing correlated gene expression patterns
Multiple Regularizers: Supports beta-VAE, DIP-VAE, beta-TC-VAE, and InfoVAE loss formulations
Single-Cell Optimized: Uses negative binomial likelihood for count data and learnable dispersion
Easy to Use: Both Python API and command-line interface
GPU Accelerated: Automatic CUDA detection for fast training
Installation

pip install iVAE
This repository is hosted at iVAE GitHub Repository.
Quick Start (Python API)
import scanpy as sc
import iVAE
# Load your single-cell data
adata = sc.read_h5ad('data.h5ad')
# Initialize and train iVAE
model = iVAE.agent(
adata,
layer='counts',
latent_dim=10,
i_dim=2,
irecon=1.0,
beta=1.0,
epochs=1000
).fit()
# Extract representations
iembed = model.get_iembed() # Interpretative embedding (n_cells x i_dim)
latent = model.get_latent() # Full latent space (n_cells x latent_dim)
# Add to AnnData for downstream analysis
adata.obsm['X_iVAE'] = iembed
adata.obsm['X_latent'] = latent
Quick Start (Command Line)
iVAE --epochs 1000 --layer counts --irecon 1.0 --data_path data.h5ad --output_dir iVAE_output
CLI Parameters
--epochs: Number of training epochs (default: 1000)--layer: Layer to use from the AnnData object (default: ‘counts’)--percent: Fraction of cells per mini-batch (default: 0.01)--irecon: Interpretative reconstruction loss weight (default: 0.0)--beta: KL divergence weight (default: 1.0)--dip: DIP loss weight (default: 0.0)--tc: Total correlation loss weight (default: 0.0)--info: InfoVAE MMD loss weight (default: 0.0)--hidden_dim: Hidden layer dimension (default: 128)--latent_dim: Latent space dimension (default: 10)--i_dim: Interpretative dimension (default: 2)--lr: Learning rate (default: 1e-4)--data_path: Path to input h5ad file (default: ‘data.h5ad’)--output_dir: Output directory (default: ‘iVAE_output’)
Output Files
After training, the following files are saved to the output directory:
iembed.npy: Interpretative embedding fromget_iembed()latent.npy: Full latent representation fromget_latent()
import numpy as np
iembed = np.load('iVAE_output/iembed.npy')
latent = np.load('iVAE_output/latent.npy')
Advanced Usage
DIP-VAE regularization (disentangled latent factors):
model = iVAE.agent(adata, layer='counts', dip=10.0, irecon=1.0).fit()
Beta-TC-VAE (total correlation decomposition):
model = iVAE.agent(adata, layer='counts', tc=5.0, irecon=1.0).fit()
InfoVAE (MMD-based regularization):
model = iVAE.agent(adata, layer='counts', info=1.0, irecon=1.0).fit()
License

This project is licensed under the MIT License.
Citation
Fu, Z., Chen, C., Wang, S., Wang, J., & Chen, S. (2025). iVAE: Interpretable Variational Autoencoder for Enhancing Clustering and Revealing Latent Biological Structure in Single-Cell Transcriptomics. BMC Biology. doi:10.1186/s12915-025-02315-7
Contact
For questions or issues, please contact Zeyu Fu at fuzeyu99@126.com.